สำหรับเรื่องนี้ผมจะขออธิบายของ Population pharmacokinetic model หรือแบบจำลองทางเภสัชจลนศาสตร์ของประชากรในเบื้องต้นซึ่ง หมายความว่า เราจะอธิบายลักษณะของเภสัชจลนศาสตร์ ของคนทั่วๆ ไป โดยที่เราสามารถจะอนุมานได้ว่า คนส่วนใหญ่ของกลุ่มประชากรนั้นๆ จะต้องมีคุณสมบัติทาง เภสัชจลนศาสตร์แบบนั้นด้วย
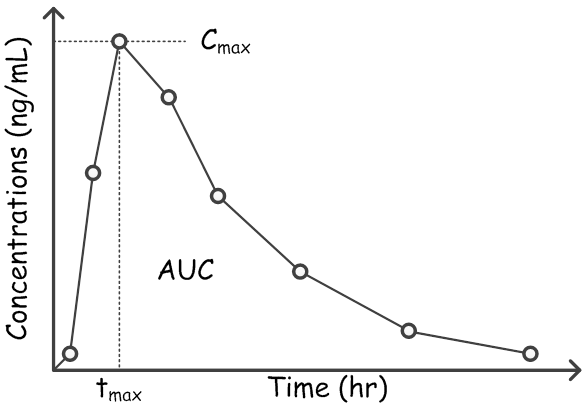
ก่อนที่จะกล่าวถึงเภสัชจลนศาสตร์ระดับประชากร ก็ต้องกล่าวถึงเภสัชจลนศาสตร์ของคนแต่ละคนก่อน หมายความว่า สมมติว่าเราสามารถสังเกต ค่าความเข้มข้นของยากับเวลา หลังจากที่กินยาเข้าไปของคนหนึ่งๆ มา เราก็สามารถเอามาพลอตกราฟ แล้วก็หาว่าคนคนนั้น มีค่าพารามิเตอร์ต่างๆ ทางเภสัชจลนศาสตร์ เป็นเช่นไร ยกตัวอย่างเช่น ความเข้มข้นของยาในเลือดที่มากที่สุด
ซึ่งวิธีแบบนี้ เป็นวิธีบอกเบื้องต้นว่า ค่าพารามิเตอร์ทางเภสัชจลนศาสตร์ ของประชากรทั้งกลุ่มเป็นอย่างไร หรือเป็นเพียงแค่การอธิบายว่าโดยสรุปแล้ว ค่าเฉลี่ยของพารามิเตอร์แต่ละตัวเป็นเช่นไร แต่อย่างไรก็ดีเราไม่สามรถที่จะสร้างแบบจำลองจากค่าเหล่านี้ได้เลย มันเป็นเพียงแค่ค่าสถิติเชิงพรรณนา (descriptive statistic) ว่าค่าพารามิเตอร์แต่ละตัวเป็นอย่างไร ซึ่งเราจะเรียกวิธีการหาค่า พารามิเตอร์ทางเภสัชจลนศาสตร์ของประชากรวิธีนี้ว่า Two-stage approach ที่เราเรียกว่า two-stage ก็คือ stage แรก คือการหาค่าพารามิเตอร์ทางเภสัชจลนศาสตร์ของแต่ละคนออกมา และ stage ที่สองก็คือ การเอาค่าพารามิเตอร์เหล่านั้น มา sum กัน หรือหาค่ากลางทางสถิติต่างๆ เพื่อที่จะอธิบายค่าพารามิเตอร์ทางเภสัชจลนศาสตร์ของประชากรที่เราสนใจ
ทีนี้สมมติว่า การศึกษาหรือการทดลองของเรา ไม่สามารถทำแบบแรกได้ อาทิ ยาที่เราสนใจมีคุณสมบัติทางเภสัชจนลศาสตร์เป็น two-compartment model ซึ่ง เป็นโมเดลที่มีพารามิเตอร์เบื้องต้นอย่างต่ำถึง 5 ตัว กล่าวคือ clearance, central volume of distribution, inter-compartmental clearance, peripheral volume of distribution, และ absorption rate constant (สำหรับค่าพารามิเตอร์อื่นๆ เช่น non-linear mixed effects modeling ในการหา
ยิ่งกว่านั้นก็คือ ถ้าเราต้องการที่จะสามารถนำเอาข้อมูลที่เราเก็บมาได้ในตอนแรก ใช้เพื่ออธิบาย หรือคาดคะเนสิ่งที่จะเกิดขึ้นในอนาคต หรือสิ่งที่เรายังไม่รู้ (extrapolation) เราไม่สามารถที่จะใช้วิธีทางสถิติอย่างง่าย หรือ two-stage approach ทำได้เลย ดังนั้นเราจำเป็นที่จะต้องใช้โมเดลในการอธิบายข้อมูลเหล่านั้น และใช้โมเดลที่ได้ ไป extrapolate ต่อ หรือเอาไป predict หรือ simulation คำถามต่างๆ ที่เราสนใจได้ ซึ่งสำหรับข้อมูลทางเภสัชจลนศาสตร์ เราก็ต้องใช้ โมเดลทางเภสัชจลนศาสตร์ของประชากร หรือเรียกว่า population pharmacokinetic model
แบบจำลองทางเภสัชจลนศาสตร์ (Pharmacokinetic model) สำหรับเรื่องนี้ ผมก็เคยได้อธิบายได้ในเบื้องต้นแล้วว่า เราจะใช้ แบบจำลองของ compartment เพื่อที่จะอธิบายระบบทางเภสัชจลนศาสตร์ โดยที่ compartment model ทุกแบบ สามารถเขียนแทนได้ด้วยสมการเชิงอนุพันธ์ (differential equation) โดยบางระบบก็สามารถแก้หาคำตอบได้ หรือที่เราเรียกว่า closed form solution หรือ analytical solution แต่อีกหลายๆ ระบบ ส่วนมากเราก็ไม่สามารถที่จะหาคำตอบแบบนั้นได้ ดังนั้น เราจำเป็นที่จะต้องใช้คำตอบแบบ numerical solution เท่านั้นในการอธิบาย โดยในที่นี้ผมจะขอยกตัวอย่างง่ายๆ ก่อนนะครับ
โดยระบบที่ผมจะยกตัวอย่างในที่นี้คือง่ายที่สุดคือ ระบบของ one-compartment model แบบที่ให้ยาทาง หลอดเลือดดำ โดยที่เราทราบสมการของระบบที่เป็น differential equation แล้ว คือ
โดยที่
และสำหรับคำตอบที่เป็น closed form solution หรือ analytical solution ของความเข้มข้นของยาที่เวลาต่างๆ ภายหลังจากที่ฉีดเข้าไป ก็คือ
และเมื่อแทนค่าสำหรับที่เวลา
และจากที่เราทราบมาแล้วจากเรื่อง พารามิเตอร์ทางเภสัชจลนศาสตร์ ว่า
โดยที่สมการนี้ จะมีพารามิเตอร์ที่เราไม่ทราบค่าอยู่สองตัว ก็คือ CL กับ V ซึ่งเราจะพยายามที่จะปรับค่า CL กับ V นี้ ให้สามารถที่จะอธิบาย ค่าความเข้มข้นของยาในกระแสเลือดที่เวลาต่างๆ ให้ได้ โดยค่าที่ดีที่สุดก็คือค่า CL กับ V ที่ทำให้ค่าความคลาดเคลื่อนระหว่างค่าจริง หรือค่าที่สังเกตได้ (observed concentrations) กับค่าที่เราคาดคะแนได้ (predicted concentrations) มีค่าน้อยที่สุด ซึ่งวิธีแบบนี้ก็เหมือนกับวิธีของการหาค่าจากสมการถดถอยเชิงเส้น หรือ linear regression แต่ว่าสิ่งที่แตกต่างก็คือ เราไม่ได้หาค่า ความชัน (slope) กับค่าจุดตัดแกน y แบบสมการเส้นตรง แต่เราหาค่า CL กับ V เพื่อนที่จะอธิบายสมการเอ็กโพแนนเชียล (exponential) ซึ่งเราก็จะเรียกมันว่าเป็น non-linear regression ก็ได้
รูปที่ 1 แสดงค่า log ของความเข้มข้นของยา กับเวลา โดยที่ความเข้มข้นของยาในร่างกายแบบนี้ อธิบายได้ด้วย one-compartment modelทีนี้ สำหรับค่าพารามิเตอร์อื่นๆ ไม่ว่าจะเป็น AUC หรือ ค่าครึ่งชีวิต เราก็สามารถหาได้จาก ค่า CL กับ V ที่หามาได้กับสูตร ที่ผมได้อธิบายไว้แล้วในเบื้องต้นก็ได้
ค่าความคลาดเคลื่อนไปจากโมเดล (residual error) ค่าความคลาดเคลื่อนไปจากโมเดล เราก็จะอธิบายด้วยผลต่างของค่าที่สังเกตได้ กับค่าที่เราคาดการณ์จากโมเดล
โดยที่เราจะถือว่าค่าความคลาดเคลื่อนของโมเดลของเราเป็นค่าคงที่ก็ได้ หรือเราอาจจะอธิบายว่าค่าความคลาดเคลื่อนเหล่านี้เป็นค่าที่แปรผันตรงกับความเข้มข้นก็ได้ หรือที่เราเรียกว่า weighted residual errors เพราะว่าโดยปกติแล้ว ค่าความคลาดเคลื่อนที่ได้จากการวัดปริมาณยาที่แต่ละค่าความเข้มข้นไม่เท่ากัน กล่าวคือ เราจะยอมรับค่าความเข้มข้นของการวัดปริมาณยาอยู่ที่ประมาณไม่เกิน ±15% หมายความว่าที่ค่าความเข้มข้นต่ำๆ ค่าความคลาดเคลื่อนก็จะต่ำไปด้วย และที่ค่าความเข้มข้นสูงๆ ค่าความคลาดเคลื่อนก็จะสูงไปด้วย โดยที่เราจะเรียกข้อมูลประเภทนี้ว่า heteroscadestic data หรือ ข้อมูลที่ค่าความคลาดเคลื่อนไม่คงที่ ส่วนข้อมูลที่ค่าความคลาดเคลื่อนคงที่เราก็จะเรียกว่า homoscadestic data ดังนั้นสำหรับข้อมูลทางเภสัชจลนศาสตร์ (กล่าวคือ ความเข้มข้นของยาในกระแสเลือด) เราก็จะอธิบายค่าความคลาดเคลื่อนของมันแบบ weithted residual errors
ทีนี้ เวลาที่เราจะอธิบายค่าความคลาดเคลื่อน เราจะพบว่าความคลาดเคลื่อนจากที่โมเดลเราคาดคะเน (prediction) จะมีการกระจายตัวแบบ normal distribution หรือสามารถเขียนเป็นสมการทางคณิตศาสตร์ได้ดังนี้
โดยที่
โมเดลประชากร (Population model) ทีนี้ เวลาที่เราจะอธิบายประชากรทั้งหมดที่เราศึกษา เราก็จะอธิบายด้วยโมเดลทางสถิติ หรือ statistical model โดยที่เราก็จะตั้งสมมติฐานเบื้องต้นเกียวกับลักษณะการกระจายตัวของพารามิเตอร์ต่างๆ ทางเภสัชจลนศาสตร์ โดยที่พารามิเตอร์ทั้งหมด เราจะนิยามให้ว่ามันมีการกระจายตัวแบบ log normal (ตามรูปที่ 2) เพราะว่าค่าพารามิเตอร์ทางเภสัชจลนศาสตร์ทุกตัวไม่สามารถมีค่าที่น้อยกว่า 0 ได้ และมีการแจกแจงแบบเบ้ขวา แต่อย่างไรก็ดี สำหรับการศึกษาทางเภสัชจลนศาสตร์ บางกรณี อาจจะจะใช้การกระจายตัวแบบอื่นๆ ในการอธิบายพารามิเตอร์ ซึ่งมันก็ขึ้นอยู่กับสมมติฐานเบื้องต้นของแต่ละการศึกษาไป โดยในที่นี้ เราจะใช้
โดยในที่นี้ เวลาที่เราจะทำให้ค่าพารามิเตอร์ทางเภสัชจลนศาสตร์ ต่างๆ มีการกระจายตัวแบบ log normal เราก็เพียงแค่ หาค่า exponantial ของมัน เราก็จะได้ค่าพารามิเตอร์ที่มีการกระจายตัวแบบ normal distribution แล้ว ซึ่งเราสามารถเขียนได้ตามสมการ
โดยในที่นี้
ในทำนองเดียวกัน สำหรับพารามิเตอร์ตัวอื่นๆ เช่น volume of distribution เราก็จะนิยามไปในทำนองเดียวกัน คือ
รูปที่ 2 แสดงการเปรียบเทียบระหว่าง Normal distribution กับ log-normal distribution โดยที่เราจะสังเกตุเห็นได้ว่าการกระจายตัวแบบ log-normal distribution เป็นการกระจายตัวแบบเบ้ขวา และมีค่าเป็นบวกเสมอ ในขณะที่ normal distribution จะมีการกระจายตัวแบบสมมาตร และค่าสามารถที่จะเป็นลบได้สรุป ทีนี้เวลาที่เราจะอธิบายข้อมูลทางเภสัชจลนศาสตร์ หรือสร้างแบบจำลองทางเภสัชจลนศาสตร์ระดับประชากร เราก็จะต้องอธิบายทุกอย่างที่กล่าวมาข้างต้นไปพร้อมๆ กันเลยคือ
Structural model คือ เราจะอธิบายว่ายาแต่ละตัว มีคุณสมบัติทาง pharmacokinetic อย่างไร กล่าวคือ อธิบายว่ามันเป็น one-compartment model, two-compartment model, มีกระบวนการดูดซึมยาอย่างไร จะเป็น zeroth-order, first-order เป็นต้นPopulational model คือ เราจะอธิบายถึงความแตกต่างของค่าพารามิเตอร์ทางเภสัชจลนศาสตร์ของประชากรแต่ละคน (inter-individual variation) ว่างแตกต่างกันอย่างไร มีการกระจายตัวอย่างไร หรือในบางกรณีเราอาจจะต้องอธิบายถึงความแตกต่างของพารามิเตอร์ทางเภสัชจลนศาสตร์ในแต่ละคนด้วย หรือ intra-individual variability โดยที่สำหรับพารามิเตอร์ทางเภสัชจลนศาสตร์บางตัว เราอาจจะหาค่าสหสัมพันธ์ (correlations) กับค่าพารามิเตอร์ของประชากรแต่ละคน (covariates) โดยเรื่องนี้ ผมขอเก็บไว้อธิบายทีหลังนะครับError model คือ การที่รวมเอาค่าความคลาดเคลื่อนของค่าที่คาดคะเนได้จากโมเดลเข้ามาร่วมการพิจารณาด้วย กล่าวคือ เวลาที่เราจะเลือกว่าโมเดลไหน ดีกว่าโมเดลไหน เราก็จะต้องดูว่าแต่ละโมเดลให้ความคลาดเคลื่อนไปเท่าไร ถ้าโมเดลไหนที่อธิบายข้อมูลแล้วมีผลค่าความคลาดเคลื่อนมาก เราก็จะเลือกใช้โมเดลที่มีค่าความคลาดเคลื่อนน้อยกว่าแต่อย่าไรก็ดี สุดท้ายแล้วเราก็ต้องหาว่าโมเดลใดสามารถอธิบายข้อมูลที่เรามีได้ดีที่สุด ดังนั้น เราก็ต้องใช้ค่า หรือปริมาณทางสถิติเข้าเปรียบเทียบ เราอาจจะเรียกช่วงนี้ว่าเป็น statistical model ไม่ว่าจะเป็นค่า objective function value, Akaike information criterion (AIC), Bayesian information criterion (BIC) หรืออื่นๆ มาอธิบายร่วมด้วย เพื่อช่วยเราตัดสินใจว่า เราจะเลือกโมเดลแบบไหนในการอธิบายข้อมูลที่เรามีอยู่ได้ดีที่สุด
/palang
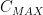


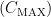 , เวลาที่ยาในเลือดจะถึงระดับมากที่สุด
, เวลาที่ยาในเลือดจะถึงระดับมากที่สุด  หรือ drug exposure
หรือ drug exposure 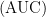 มีค่าเป็นเท่าไร แต่ถ้าเราจะหาค่าของประชากรที่เราศึกษาทั้งหมด ยกตัวอย่างเช่น เราทำการวิเคราะห์ค่าทางเภสัชจลนศาสตร์มาจากคน 50 คน เราก็สามารถที่จะหาค่าของประชากรได้โดย หาค่าสรุปจากวิธีทางสถิติง่ายๆ ได้ อาทิ เราสามารถที่จะใช้ค่าเฉลี่ย (mean) ของประชากรทั้งหมดที่เราศึกษา หรือ ค่ามัธยฐาน (median) ของประชากรที่เราศึกษา และใช้ค่าส่วนเบียงเบนมาตรฐาน หรือค่าความแปรปรวน เพื่อบอกถึงความต่างของประชากรแต่ละคน เช่นตัวอย่าง
มีค่าเป็นเท่าไร แต่ถ้าเราจะหาค่าของประชากรที่เราศึกษาทั้งหมด ยกตัวอย่างเช่น เราทำการวิเคราะห์ค่าทางเภสัชจลนศาสตร์มาจากคน 50 คน เราก็สามารถที่จะหาค่าของประชากรได้โดย หาค่าสรุปจากวิธีทางสถิติง่ายๆ ได้ อาทิ เราสามารถที่จะใช้ค่าเฉลี่ย (mean) ของประชากรทั้งหมดที่เราศึกษา หรือ ค่ามัธยฐาน (median) ของประชากรที่เราศึกษา และใช้ค่าส่วนเบียงเบนมาตรฐาน หรือค่าความแปรปรวน เพื่อบอกถึงความต่างของประชากรแต่ละคน เช่นตัวอย่าง
 หรือ
หรือ  เราสามารถหาจากพารามิเตอร์เบื้องต้นที่กล่าวมาแล้วได้) และเราทำการทดลองในกลุ่มประชากรเด็กเล็ก ซึ่งแน่นอนว่า ถ้าเราต้องการที่จะหาค่าพารามิเตอร์ทั้ง 5 ตัวนี้ของประชากรเด็กในแต่ละคนนั้น เราจำเป็นที่จะต้องมีตัวอย่างเลือด อย่างน้อย 5 ตัวอย่าง จากเวลาที่แตกต่างกัน เพื่อที่จะเอามาหาค่าพารามิเตอร์ที่เราต้องการได้ แต่อย่างที่บอก การทดลองในเด็ก เราไม่สามารถที่จะเก็บเลือดเด็กเป็นจำนวนมากอย่างที่เราต้องการได้ ดังนั้น เราจึงไม่สามารถที่จะใช้วิธี two-stage approach ในการหาอธิบายเภสัชจลนศาสตร์ประชากรได้ ซึ่งในกรณีนี้ เราต้องใช้วิธีที่ซับซ้อนขึ้นไปอีกขั้นหนึ่ง คือ non-linear mixed effects modeling ในการหา
เราสามารถหาจากพารามิเตอร์เบื้องต้นที่กล่าวมาแล้วได้) และเราทำการทดลองในกลุ่มประชากรเด็กเล็ก ซึ่งแน่นอนว่า ถ้าเราต้องการที่จะหาค่าพารามิเตอร์ทั้ง 5 ตัวนี้ของประชากรเด็กในแต่ละคนนั้น เราจำเป็นที่จะต้องมีตัวอย่างเลือด อย่างน้อย 5 ตัวอย่าง จากเวลาที่แตกต่างกัน เพื่อที่จะเอามาหาค่าพารามิเตอร์ที่เราต้องการได้ แต่อย่างที่บอก การทดลองในเด็ก เราไม่สามารถที่จะเก็บเลือดเด็กเป็นจำนวนมากอย่างที่เราต้องการได้ ดังนั้น เราจึงไม่สามารถที่จะใช้วิธี two-stage approach ในการหาอธิบายเภสัชจลนศาสตร์ประชากรได้ ซึ่งในกรณีนี้ เราต้องใช้วิธีที่ซับซ้อนขึ้นไปอีกขั้นหนึ่ง คือ non-linear mixed effects modeling ในการหา
 คือ elimination rate constant ของยา นิยามว่า
คือ elimination rate constant ของยา นิยามว่า 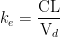 และ ที่เวลา
และ ที่เวลา 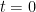 initial condition ของ compartment 1 คือ Dose ของยา
initial condition ของ compartment 1 คือ Dose ของยา 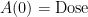

 ตามสมการ
ตามสมการ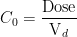
 ก็จะทำให้สมการความเข้มข้นของยาที่เวลาต่างๆ เป็น
ก็จะทำให้สมการความเข้มข้นของยาที่เวลาต่างๆ เป็น


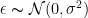
 คือค่าความคลาดเคลื่อน หรือ residual errors ซึ่งจะมีการกระจายตัวแบบ normal distribution ด้วยค่าเฉลี่ยเท่ากับ 0 และมีค่าส่วนเบียงเบนมาตรฐานเป็น
คือค่าความคลาดเคลื่อน หรือ residual errors ซึ่งจะมีการกระจายตัวแบบ normal distribution ด้วยค่าเฉลี่ยเท่ากับ 0 และมีค่าส่วนเบียงเบนมาตรฐานเป็น 
 แทนค่าความแปรปรวนของแต่ละคนที่แตกต่างจากค่ากลางของข้อมูลนะครับ โดยที่ ค่า
แทนค่าความแปรปรวนของแต่ละคนที่แตกต่างจากค่ากลางของข้อมูลนะครับ โดยที่ ค่า  ตามสมการ
ตามสมการ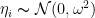

 เป็นค่า clearance ของคนใดคนหนึ่งในกลุ่มประชากรที่เราศึกษา
เป็นค่า clearance ของคนใดคนหนึ่งในกลุ่มประชากรที่เราศึกษา  คือค่า clearance เฉลี่ยของประชากร หรือจะบอกว่าเป็นค่า clearance ที่เป็นตัวแทนของคนส่วนมากในประชากรนั้นๆ และค่า
คือค่า clearance เฉลี่ยของประชากร หรือจะบอกว่าเป็นค่า clearance ที่เป็นตัวแทนของคนส่วนมากในประชากรนั้นๆ และค่า  คือค่าความแปรปรวนของประชากรนั้นๆ หรือปริมาณที่บ่งบอกถึงความไม่เหมือนกันของคนแต่ละคน (inter-individual variability)
คือค่าความแปรปรวนของประชากรนั้นๆ หรือปริมาณที่บ่งบอกถึงความไม่เหมือนกันของคนแต่ละคน (inter-individual variability)

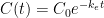

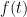 ไปเป็นฟังก์ชันใน Laplace space หรือ
ไปเป็นฟังก์ชันใน Laplace space หรือ  ตามตาราง Laplace transform ข้างล่าง แล้วก็แก้สมการแบบพีชคณิตใน Laplace space แล้วสุดท้ายก็แปลงสมการกลับมาเป็น time space เหมือนเดิม โดยในที่นี้ ผมจะขอยกตัวอย่างซักสองสามตัวอย่างนะครับ โดยตัวอย่างทั้งหมดในที่นี้ จะเป็นตัวอย่างของระบบทาง
ตามตาราง Laplace transform ข้างล่าง แล้วก็แก้สมการแบบพีชคณิตใน Laplace space แล้วสุดท้ายก็แปลงสมการกลับมาเป็น time space เหมือนเดิม โดยในที่นี้ ผมจะขอยกตัวอย่างซักสองสามตัวอย่างนะครับ โดยตัวอย่างทั้งหมดในที่นี้ จะเป็นตัวอย่างของระบบทาง


![\dfrac{\text{d}}{\text{dt}} \text{A}_i = \text{[Rate in]} - \text{[Rate out]}](https://s0.wp.com/latex.php?latex=%5Cdfrac%7B%5Ctext%7Bd%7D%7D%7B%5Ctext%7Bdt%7D%7D+%5Ctext%7BA%7D_i+%3D+%5Ctext%7B%5BRate+in%5D%7D+-+%5Ctext%7B%5BRate+out%5D%7D&bg=ffffff&fg=000000&s=0&c=20201002)
 ซึ่งค่า
ซึ่งค่า  คือค่า rate constant สำหรับยาที่ไหลจาก compartment ที่
คือค่า rate constant สำหรับยาที่ไหลจาก compartment ที่ 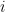 ไปยัง compartment ที่
ไปยัง compartment ที่ 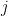 และค่า
และค่า  คือปริมาณยาที่อยู่ใน compartment ที่
คือปริมาณยาที่อยู่ใน compartment ที่  ตามรูปที่ 1
ตามรูปที่ 1
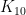 คือค่า elimination rate constant หรือ ค่าคงตัวของอัตราการกำจัดยาออกจากร่างกาย
คือค่า elimination rate constant หรือ ค่าคงตัวของอัตราการกำจัดยาออกจากร่างกาย หรือ fraction of gut absorption
หรือ fraction of gut absorption ดังนั้น สัดส่วนของยาที่จะสามารถผ่านตับไปได้คือ
ดังนั้น สัดส่วนของยาที่จะสามารถผ่านตับไปได้คือ 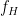 ซึ่งนิยามว่าเป็น
ซึ่งนิยามว่าเป็น 
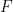 คือสัดส่วนของยาที่เหลืออยู่ หรือสัดส่วนของยาที่สามารถเข้าสู่ systemic circulation ได้ นั่นก็คือ ผลคูณของสัดส่วนของยาที่สามารถผ่านผนังทางเดินอาหารมาได้ (
คือสัดส่วนของยาที่เหลืออยู่ หรือสัดส่วนของยาที่สามารถเข้าสู่ systemic circulation ได้ นั่นก็คือ ผลคูณของสัดส่วนของยาที่สามารถผ่านผนังทางเดินอาหารมาได้ (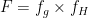

 หรือ ค่าความเข้มข้นของยาที่มากที่สุด (maximum concentrations) ในกระแสเลือดของเราภายหลังจากที่เรากินยาเข้าไป (หรืออาจะเป็นกรณีอื่นๆ สำหรับการให้ยานอกหลอดเลือดดำ, extravascular administration, อาทิ intra-muscular administration, intra-rectal administrations เป็นต้น) สำหรับการให้ยาทางหลอดเลือดดำหรือ intra-venous administration นั้น ค่าความเข้มข้นของยาที่มาที่สุด จะเกิดขึ้นตอนเวลาเป็นศูนย์ คือ ไม่มีช่วง absorption phase เลย แต่ถ้าเป็น intra-venous bolus infusion ค่าความเข้มข้นของยาที่มาที่สุด จะเกิดขึ้นตอนที่ สิ้นสุด การ infuse ยาเข้าหลอดเลือดดำครับ
หรือ ค่าความเข้มข้นของยาที่มากที่สุด (maximum concentrations) ในกระแสเลือดของเราภายหลังจากที่เรากินยาเข้าไป (หรืออาจะเป็นกรณีอื่นๆ สำหรับการให้ยานอกหลอดเลือดดำ, extravascular administration, อาทิ intra-muscular administration, intra-rectal administrations เป็นต้น) สำหรับการให้ยาทางหลอดเลือดดำหรือ intra-venous administration นั้น ค่าความเข้มข้นของยาที่มาที่สุด จะเกิดขึ้นตอนเวลาเป็นศูนย์ คือ ไม่มีช่วง absorption phase เลย แต่ถ้าเป็น intra-venous bolus infusion ค่าความเข้มข้นของยาที่มาที่สุด จะเกิดขึ้นตอนที่ สิ้นสุด การ infuse ยาเข้าหลอดเลือดดำครับ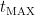 คือ เวลาหลังจากที่เรากินยาเข้าไปแล้วปริมาณยาในกระแสเลือดถึงระดับ
คือ เวลาหลังจากที่เรากินยาเข้าไปแล้วปริมาณยาในกระแสเลือดถึงระดับ  แต่ถ้าเป็น intra-venous infusion ค่า
แต่ถ้าเป็น intra-venous infusion ค่า